
目录
前言:我们需要更好的相关性热力图
在 Python 中我们日常分析数据的过程当中经常需要对数据进行相关性分析,相关性热力图(Correlation Heatmap)是我们经常使用的一种工具。通过相关性热力图,我们可以通过为相关性不同的数据使用不同深浅的不同颜色进行标记,从而直观地观察两两数据序列之间的相关性情况——这将有助于我们进一步的数据分析和处理,比如数据的回归分析等。
这其中最常见的工具就是由 Seaborn 工具包提供的 sns.heatmap(),处理方法的原理相当于先取得变量序列的相关性矩阵,然后直接对相关性矩阵绘制矩阵热图。
然而最近在学习了 R 语言之后,使用 corrplot 包可以绘制出更加华丽、全面、直观的相关性图,相比较之下就觉得 Seaborn 提供的热图并不令人满意。因此本文介绍一种新的更好的相关图的绘制方法,来自 Biokit 工具包中的 biokit.viz.Corrplot() 类,并介绍其使用。
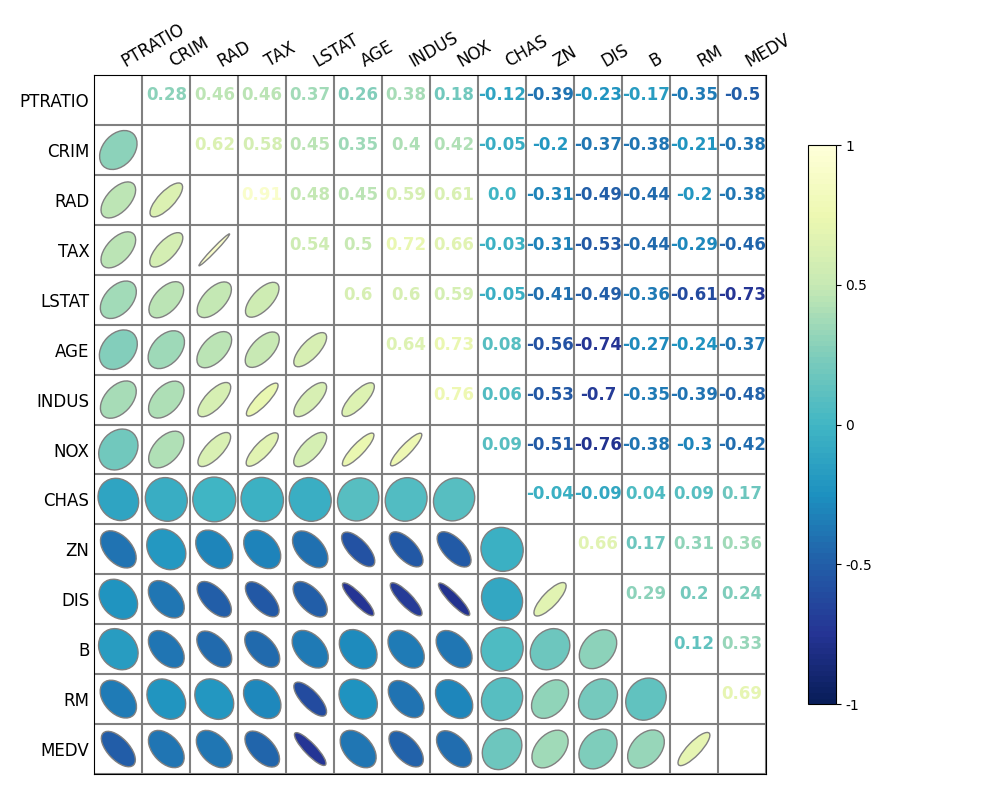
废话不多说,先上图展示:

- (咱就说我们平时写论文或者分析报告的时候,把这个图往上面一放是不是高端大气上档次?)
对比 Python Seaborn 与 R corrplot
传统的 Seaborn 相关性热力图
下面展示了一个简单的使用示例:
# 导入需要用到的库
import pandas as pd
import matplotlib.pyplot as plt
import seaborn as sns
# 读取数据,这里以著名的波士顿数据集举例
data = pd.read_csv('./data/boston.csv')
# 绘制热图
fig, ax = plt.subplots(figsize=(10, 8))
sns.heatmap(
data.corr(), annot=True, cmap='coolwarm', ax=ax)
fig.savefig('../image/py_heatmap.png')
上述的代码绘制效果如下图所示:

从上图中可以看出传统的 Seaborn 的热图存在一些问题:
- 各个字段按顺序输出,不同相关性的单元格混排,难以一眼看出变量之间的显著相关关系
- 即使选择了对比度更高的,Colormap 仍然难以一眼找到具有较高相关性的变量
- 三角图的绘制需要设置和使用 mask 参数,代码书写复杂度瞬间翻倍
R 语言中的相关性热力图
相比较之下,如下是通过 R 语言绘制的相关性热图的示例:
### 导入工具包 ###
library('corrplot')
### 读取数据 ###
dat <- read.csv('./data/boston.csv')
### 计算取得相关性及相关性显著性 ###
corr <- cor(dat)
corrl <- cor.mtest(dat)
### 绘图 ###
corrplot.mixed(
corr, order = 'AOE',
lower = 'number', upper = 'ellipse',
p.mat = corrl$p, insig = 'pch', pch = 4,
tl.col = 'black', tl.pos = 'lt',
number.cex = 0.7, # 调整数字大小
)
关于 R 语言中 Corrplot 的使用方法,可以参考文章 An Introduction to corrplot Package,从原文中可以看见 R 的 Corrplot 包提供了大量自由度极高的参数选项对绘制的相关性热图进行调整。
绘制效果如下图所示:

从上图可以看出,与 Seaborn 相比,R 语言的相关性热力图具有以下几个显著的优势:
- 允许使用椭圆形、不同大小的圆或者方片乃至小饼图结合颜色标记来标记每个变量之间的相关性,更加直观、清晰。
- 允许数值和形状分开分别绘制在图的左下角和右上角,丰富图的内容。
- 对于未通过显著性检验的相关性矩阵元素,使用散点标记划去(图中的叉)。
显然我们希望在 Python 中也能像这样轻松第绘制更加美观和华丽的相关性热力图。
关于 Biokit
简介
BioKit 一套专门用于生物信息学、数据可视化(BioKit.viz)、在线生物数据访问(例如 UniProt,得益于生物服务的 NCBI)的工具。它还包含与数据分析相关的更高级的工具(例如 biokit.stats)。由于R在生物信息学中很常见,Biokit 还提供了一个方便的模块来在 Python 脚本或 shell 中运行 R(biokit.rtools 模块)。
文档手册在 biokit.readthedocs.io,详情请自行查看。
库的安装
Biokit 的安装在原生的 Python 和 Anaconda 中略有不同,按照官方给出的安装方式,在 Python 中:
pip install biokit
如果在使用 Conda,则官方手册和代码仓库的 README.md 文件中给出的安装方式互不相同。其原因……额,咱也不知道咱也不敢问:
conda install bioconda # 根据官方仓库 README.md
conda install biokit # 根据手册
不过我会推荐大家从源码构建安装 Biokit,具体的原因我会在下文详述。
相关性热图的绘制
基本使用方法
Biokit 中的热图是以一个类的形式定义的,具体的定义形式:
biokit.viz.Corrplot(data, na=0)
这里初始化方法传入两个参数:na 是一个用于替换数据中的缺失值的默认值,也就是说如果数据里面有缺失值就会被自动替换为这个值。不言而喻;
这个 data 则很有意思,我翻译了官网上对于这个变量的介绍:
输入可以是 DataFrame(Pandas)、List(python)或 numpy 矩阵。但是,请注意,值必须介于 -1 和 1 之间。如果不是, 或者如果矩阵(或列表列表)不是方阵,则相关性为其自动计算的结果。数据或计算的相关性存储在
df属性中。
也就是说,在 Biokit 中,传进的 data 参数可以是有待计算相关性的原始数据,也可以是相关性矩阵。如果传入前者,方法会为你计算相关性系数。
完成初始化之后,需要调用 .plot() 方法来绘图。其中,关于绘制图片的方法 biokit.viz.Corrplot.plot() 定义形式如下:
def plot(
self, fig=None, grid=True, rotation=30,
lower=None, upper=None, shrink=0.9, facecolor="white",
colorbar=True, label_color="black", fontsize="small",
edgecolor="black", method="ellipse", order_method="complete",
order_metric="euclidean", cmap=None, ax=None,
binarise_color=False):
详述一些绘图参数的问题及细节
对于每个参数的含义的说明,如下表所示:
| 名称 | 默认值 | 含义 |
|---|---|---|
fig |
None |
一个 matplotlib 图对象,用于绘制热力图。默认情况下会自动创建,也可以在使用的时候具体指定。 |
grid |
True |
添加网格(默认为灰色)。您可以将其设置为False或颜色。 |
rotation |
30 |
在y轴上旋转标签的角度 |
lower |
None |
设置左下角部分的热力图的绘制方法,可选的参数有 'ellipse','square','rectangle' 以及'color','text','circle','number' 和 'pie',绘制效果即字面意思所示,本文的读者可自行尝试每一种绘制效果。 |
upper |
None |
设置右上上角部分的热力图的绘制方法,可选参数同 lower,此不复赘述。 |
shrink |
0.9 |
符号使用每个小方格的最大空间(百分比)。如果提供负值,则取绝对值;如果大于 1,符号将与边框方格重叠。 |
facecolor |
"white" |
图整体背景的颜色(默认为白色)。 |
colorbar |
True |
添加颜色条(默认为True)。 |
label_color |
"black" |
轴标签的颜色(默认为黑色)。 |
fontsize |
"small" |
字体的大小,默认为“small”。可以直接用数字控制大小。 |
edgecolor |
"black" |
代码文档中未曾提及但是确实存在的参数选项,目测其功能是指定绘制矩形、椭圆、圆等图形时图形边缘的颜色。。 |
method |
"ellipse" |
在没有指定 lower 或者 upper 参数时生效, |
order_method |
"complete" |
使用方法对相关性矩阵的行和列重新排序,使得相似的变量被聚集在一起。这种排序可以帮助你更清晰地观察和理解数据的相关性模式。 |
order_metric |
"euclidean" |
用于计算距离的度量标准。下文详述。 |
cmap |
None |
matplotlib 或 colormap 包中的有效的 Colormap(例如。'jet' 或 'copper')。默认为红色/白色/蓝色。 |
ax |
None |
一个标准的 matplotlib 坐标轴对象。不指定则自动创建。 |
binarise_color |
False |
又是一个代码文档中没说过的参数。目测其功能是将颜色值二值化(binarize)。具体来说,如果 binarise_color 被设置为 True,颜色值将被转换为二进制值。换句话说,简化颜色表示,仅使用两种颜色来表示正负相关性。 |
对于上表中的部分参数,我这里需要给出一些详细的说明:
我写不动了,这段内容我让 Chat 姐帮我写了。请读者们体谅一下。
关于 order_method 参数
order_method 参数决定了用于层次聚类的链接方法。这些方法定义了如何计算聚类之间的距离。常见的链接方法包括:
- ‘single’: 最近邻法。两个聚类之间的距离由最近的一对点决定。
- ‘complete’: 最远邻法。两个聚类之间的距离由最远的一对点决定。
- ‘average’: 平均法。两个聚类之间的距离由所有点对之间的平均距离决定。
- ‘centroid’: 质心法。两个聚类之间的距离由它们质心之间的距离决定。
- ‘median’: 中位数法。计算两个聚类的中位数质心之间的距离。
- ‘ward’: Ward法。最小化每个聚类的平方和误差。
这些方法是通过 scipy.cluster.hierarchy.linkage 函数实现的。选择不同的方法会影响聚类结果,从而影响相关性矩阵的排序。
关于 order_metric 参数
order_metric 参数决定了用于计算距离的度量标准。常见的度量标准包括:
- ‘euclidean’: 欧几里得距离。
- ‘cityblock’: 曼哈顿距离(城市街区距离)。
- ‘cosine’: 余弦距离。
- ‘hamming’: 汉明距离。
- ‘jaccard’: 杰卡德距离。
这些度量标准是通过 scipy.spatial.distance 或 scipy.cluster.hierarchy 提供的。选择不同的距离度量会影响聚类计算的结果。
在使用中,order_method 和 order_metric 会对相关性矩阵的行和列重新排序,使得相似的变量被聚集在一起。这种排序可以帮助你更清晰地观察和理解数据的相关性模式。为了方便读者理解,我们这里给出一段示例代码——例如,如果你使用 ‘complete’ 方法和 ‘euclidean’ 距离,对应的代码如下:
Y = self.linkage(
self.df,
method=order_method,
metric=order_metric)
ind1 = hierarchy.fcluster(
Y, 0.7 * max(Y[:, 2]), "distance")
Z = hierarchy.dendrogram(Y, no_plot=True)
idx1 = Z["leaves"]
cor2 = self.df.iloc[idx1, idx1]
这段代码首先计算相关性矩阵的层次聚类,然后根据聚类结果对矩阵的行和列进行重新排序。self.linkage 函数调用了 scipy.cluster.hierarchy.linkage,hierarchy.fcluster 和 hierarchy.dendrogram 分别用于获取聚类结果和绘制树状图。
关于 cmap 参数
Biokit 源代码中应用 camp 参数的方法是:
self.cm = cmap_builder(cmap)
所以你可以使用如下的指定 colormap 的方法:
- 指定 colormap 的名称:
cmap='viridis' - 指定三种具体颜色的过渡:
cmap=['#2F7FC1', '#FFFFFF', '#D8383A']
不要使用 如下的方法指定 colormap,否则将会报错:
# 不要这样做!
import matplotlib.pyplot as plt
from
cmap = plt.cm.get_cmap('inferno') # 获取cmap对象
data = ...
... # 中间代码略
corrp = Corrplot(data)
corrp.plot(
cmap=cmap
)
改进 BioKit 中 Corrplot 对 colormap 的支持
事情的起因
回到本文最开头的那张图片,绘制这张图片我使用了如下的代码:
import pandas as pd
import matplotlib.pyplot as plt
from biokit.viz import Corrplot
data = pd.read_csv('./data/boston.csv')
fig, ax = plt.subplots(figsize=(10, 8))
datacorr = Corrplot(data)
datacorr.plot(
ax=ax,
lower='ellipse', upper='text',
cmap='YlGnBu_r', edgecolor='grey',
fontsize=12, order_method='centroid')
如果读者们运行我给出的这段代码,就会发现图片绘制出来的效果和本文中所展示的有所不同——为什么右上半个三角中的数字只有红蓝两种颜色,而本文中展示的绘制效果则是叠加了 viridis Colormap 的效果呢?
实际上,原生的 Biokit 中 Corrplot 在使用 method='number' 的时候确实不支持任意 Colormap,只支持红蓝双色。参考 Biokit 在 Github 上的源代码仓库 biokit/biokit 中的 Jupyter Notebook biokit/viz/corrplot 中的说明:
c.plot(method='text', fontsize=8, colorbar=False) # only red to blue colormap is implemented so far

但是仔细查看源代码之后,我发现想要修复这个问题使 .plot(method='text') 支持 Colormap 其实很简单。参考 stackoverflow 上的提问 Getting individual colors from a color map in matplotlib,我发现 matplotlib 中有个 Normalize 类能够将 colormap 对象映射到任何数值范围内:
matplotlib.colors.Normalize(vmin=vmin, vmax=vmax)
我们知道相关性是一个从 -1 到 1 之间的数值,因此只需要将用户指定的 Colormap 映射到这个区间范围,再根据每个相关性矩阵元素的值从标准化之后的 Colormap 中选取颜色就好了。具体的实现方法:
Biokit 中原始的这段代码为:
...
elif method in ['number', 'text']:
from easydev import precision
if d<0:
edgecolor = 'red'
elif d>=0:
edgecolor = 'blue'
ax.text(x,y, precision(d, 2), color=edgecolor,
fontsize=self.fontsize, horizontalalignment='center',
weight='bold', alpha=max(0.5, d_abs))
# withdash=False)
...
我给修改成了:
...
elif method in ['number', 'text']:
from easydev import precision
# if d<0:
# edgecolor = 'blue'
# elif d>=0:
# edgecolor = 'red'
# Instantiate a
# `matplotlib.colors.Normalize()` object
# as `color_norm`
color_norm = Normalize(
# the range of correlation coefficient
vmin=-1.0,
vmax=1.0)
# select colors for each `d`
# with normalized colormap
edgecolor = self.cm(
color_norm(d))
# plot the label with the color
ax.text(
x,
y,
precision(d, 2),
color=edgecolor,
fontsize=self.fontsize,
horizontalalignment='center',
weight='bold',
# alpha=max(0.5, d_abs)
# withdash=False)
) # alpha is no longer needed
...
我当时觉得这个改动还蛮好的,所以大概是在这篇博客写完之前的上个星期,我向原仓库提交了 Feature: A Method to Apply colormap to corrplot while Using method=’number’ #78,结果原作者一直不回复我。后来我干脆自己动手,又提交了 Pull Request for Issue #78: Apply Colormap to Corrplot #79。后来我看了一下 Biokit 仓库现在的情况——好家伙,上次更新都已经是 3 年前了,不知道为什么这个仓库自那之后便一直没有再更新过。
所以读者们如果想要使用像本文开头所展示的那样带颜色的 text Corrplot,可以从 Github 上拉取 我 fork 并修改过源码的 repo,然后在本地通过源码构建安装。
拉取 Biokit 源码并在本地构建
如果您的计算机环境上已经配置过 Git 工具则可以使用下面的命令拉取项目到本地:
git clone https://github.com/GitHubonline1396529/biokit.git
进入本地项目文件夹并构建安装源码:
cd ./biokit
pip install .
注意!不要再使用 python -u ./setup.py 了! 这种安装方式已经严重过时,被弃用了。(居然到现在为止 CSDN 上还有那么多博文在建议这样构建源码,每天都有一大堆新手跑过来问我。我还能说什么?)
总结和补充
Biokit 是一个功能强大的库,尽管本文仅介绍其热力图的绘制,但是实际上这个工具包的功能非常的多,有待读者们发现。但是中文互联网上对于 Biokit 的介绍很少,我直接通过 Bing 搜索找到的也只有腾讯云社区上的 BioKit!让你用Python也可以轻松绘制矩阵热力图… 这一篇。
不知道原项目后面会不会更新,但是这么好的项目沉了也是蛮可惜的。但是凭我自学 Python 两年半的功夫肯定是更新不动这么大的一个项目的。
我也相当于是在这里问问有没有人愿意提供帮助吧。
1.本站内容仅供参考,不作为任何法律依据。用户在使用本站内容时,应自行判断其真实性、准确性和完整性,并承担相应风险。
2.本站部分内容来源于互联网,仅用于交流学习研究知识,若侵犯了您的合法权益,请及时邮件或站内私信与本站联系,我们将尽快予以处理。
3.本文采用知识共享 署名4.0国际许可协议 [BY-NC-SA] 进行授权
4.根据《计算机软件保护条例》第十七条规定“为了学习和研究软件内含的设计思想和原理,通过安装、显示、传输或者存储软件等方式使用软件的,可以不经软件著作权人许可,不向其支付报酬。”您需知晓本站所有内容资源均来源于网络,仅供用户交流学习与研究使用,版权归属原版权方所有,版权争议与本站无关,用户本人下载后不能用作商业或非法用途,需在24个小时之内从您的电脑中彻底删除上述内容,否则后果均由用户承担责任;如果您访问和下载此文件,表示您同意只将此文件用于参考、学习而非其他用途,否则一切后果请您自行承担,如果您喜欢该程序,请支持正版软件,购买注册,得到更好的正版服务。
5.本站是非经营性个人站点,所有软件信息均来自网络,所有资源仅供学习参考研究目的,并不贩卖软件,不存在任何商业目的及用途








暂无评论内容